Factors Shaping Study Timelines: Screenshots, Localized Content, and the Complex Role of IRB Submission Timing

Imagine a time when Sponsors and CROs can manage the study submission process without any dependencies on external parties. Having part of a process dependent on external parties adds risk and undue pressure on all parties and ultimately impacts patients who are waiting for the treatments that are to be tested.
In this scenario, the external parties are eCOA vendors, and the reality is that eCOA vendors are asked to develop studies to get to a point where they can generate materials that are used to support regulatory submissions. This means that the development work is done under risk, as there is always the possibility that changes have to be made. You would never build a house without approved designs, so why would you do that with your clinical trial?
Clinical study development relies on an understanding of the final deliverable, and vendors will contract and build to that deliverable. Any changes in that overall deliverable then incur change orders and delays in study deployment, impacting FPI and delaying study timelines, ultimately impacting future revenue streams from the treatment being evaluated.
Why do we have this interdependency?
Diagram 1:

When it comes to clinical research, study approvals fall into two parts: firstly, we have protocol approval, and once that is obtained, we have Institutional Review Board (IRB in the US)/Ethics (for the rest of the world) approval, which enables the clinical trial to be deployed within a given region. The ideal scenario referenced above would follow ROW A (for the US) and ROW C (for the EU) in Diagram 1.
So, is it right that vendors should start study development when a protocol has not been approved—at least by one regulatory agency? Are customers willing to accept the costs and delays associated with protocol amendments during the study deployment phase? This is controllable by ensuring the protocol has been reviewed and, ideally, approved by the regulators (the FDA or EMA under a Part 1 submission under the EU Clinical Trials Regulation). That way, you have confidence in the overall design of the study. This part is straight-forward. This is ROW B in Diagram 1.
However, when it comes to the second part of the submissions process—IRB/Ethics approval—this is where the main challenges occur. Why are IRB/Ethics submissions a challenge for vendors? Well, it’s because the expectation is that vendors will provide screenshots and localized, translated application (app) content to support the submission process. Generating screenshots and localized app content may well be actioned by the ‘press of a button, but to get that button ready, the study must be built and localized. If the study is not finalized (at a stage where everything is agreed upon, from study design to patient-facing content), how can vendor-generated screenshots and localized app content be deemed finalized? Once the study gets IRB/Ethics approval, that’s it; any updates to study content and flow cannot be applied without further IRB/Ethics review and further delays (see Diagram 2 below, Launch Process Dependent on Screenshots).
Let’s hit pause here and ask the question, "Why are we doing this?"
Believe it or not, there was a time when clinical trials were done on paper; going electronic was something new, and there was reticence and misunderstanding around what ‘going electronic’ meant. To address this, the industry (as it was then) decided to promote understanding within the IRB/Ethics community of the ‘minimal impact’ on patients of going electronic by supplementing IRB/Ethics submissions with screenshots of the electronic implementation. As clinical research expanded globally and electronic adoption took off, this was extended to include localised app content screenshots for regional Ethics boards. Hence, as an industry, we created the expectation with regards to screenshots and localized app content with Sponsors, CROS and IRB/Ethics boards.
But is the expectation valid?
Globally, regulatory authorities abide by ICH E6 Good Clinical Practices (GCP) standards when assessing clinical trial materials. This predates going electronic and focuses on informed consent and written information related to recruitment, referred to as ‘Patient Facing Materials’. Our industry interprets the GCP Guidance to apply to ‘any’ written information that is presented to the patient. Which is best summarized by Gartel et al in 2020(1) who said, "Requiring screenshots of translations of all diaries and questionnaires into local languages is an expansive reading of ICH E6 Guidance".
Diagram 2:
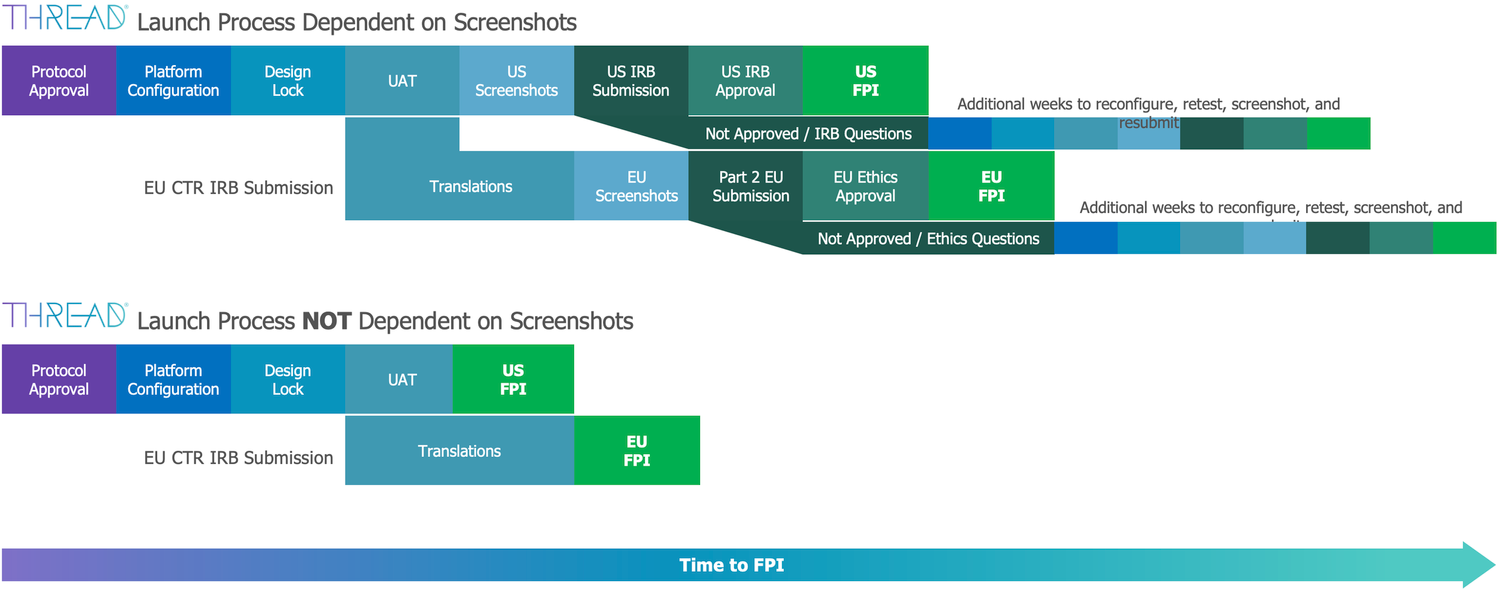
As we all know, when expectations have been set, it takes a seismic shift to hit reset. In our industry, change is a slow process, and shifts are driven by regulation. From a US perspective, the change was initiated by the IRB/Ethics boards themselves. In 2022, WCG and Advarra stated that they did not expect to see screenshots of standardized eCOA as part of their submissions. They would, at best, accept paper versions of the COA. This aligns with the COA form as part of the protocol submission (which, remember, happens before IRB/Ethics approval). Hence, IRB/Ethics boards can then take a step back and focus on the true intent of the ICH guidance and focus on consent and recruitment materials.
This approach is great from a US perspective as it means FDA and then IRB approvals can occur now without the dependency on a vendor – See Diagram 1, ROW A.
However, clinical research is global; running studies in other regions, such as the EU, has its own challenges, namely around the breadth of languages and the fact that prior to the EU Clinical Trials Regulation (CTR) coming into force on 31st January 2022, each national or regional authority had the freedom to issue specific guidance and regulation for studies conducted under their jurisdiction. This meant that Sponsors and CROs had to navigate regional regulations and multiple within-country Ethics committees, and for vendors, it meant having to produce multiple screenshots and localized app content in preparation for each Ethics submission.
The CTR standardizes the European Union submission process, and in essence, it follows the US model, where there is a central protocol approval process (Part 1) that applies to all member states, followed by member state, country level centralized Ethics approvals (Part 2). The Part 1 process includes not only the protocol but also the COA that are to be collected. The regulation clearly states that the COA submission should be paper versions and be in English, so NO screenshots or localised app content. This makes sense as the Part 1 submission, if approved, is valid for 2 years, and many sponsors will get Part 1 approval well in advance of selecting an eCOA vendor. Therefore, how will a sponsor know at the point of the Part 1 submission what the electronic screenshots will look like and what the localized content should be? Sometimes Part 1 submission occurs at the start of the study build, but as there is no requirement for screenshots and localized app content, it removes the time pressures from the study teams and allows for the development of the study without distractions.
Under Part 2, the Ethics approval process is now a centralised process within each member state. Where each member state has a single point for Ethics approval. This helps standardise Ethics board interactions, but more importantly, the CTR defines what is expected to be included to support the Part 2 submissions process.
The Part 2 process mentions the familiar term "Patient Facing Materials", which again could lead to a misinterpretation as to the exact definition. However, this has been raised with the European Medicines Agency (as part of the CTR question and answer process), and whilst the answer is not initially definitive, if you trace the responses through the CTR regulation, ultimately you get to a definition of ‘Patient Facing Materials", which is that it relates to consent and recruitment materials (back to the original ICH definition). Therefore, for Part 2 submissions, patient facing materials are NOT eCOA screenshots or localized app materials. This then reflects the ‘Launch Process NOT Dependent on Screenshots in Diagram 2 above, which means within the US and EU we can deliver studies to patients faster.
On the flipside, if you include the eCOA in your patient facing recruitment and consenting processes, then yes, it is reasonable to expect screenshots and localized app content to be included in the Part 2 process.
So, in summary, what are we saying?
- As an industry, we have created chaos by misinterpreting the guidance for screenshot submissions.
- Maintaining the norm has an impact on trial timelines and preparedness, with a dependency being set on the creation of materials for Part 2 submission, which puts undue pressure on all parties involved in the study development process and creates necessary coasts and delays.
- It is on us (the industry) to drive the necessary changes with our customers and partners to reset and enable more agile implementation experiences that STILL meet regulatory requirements.
For more information, read our recent report that covers this topic extensively.
References: